Was ist der Unterschied zwischen Tay-Sachs und Sandhoff-Krankheit
Der Schlüsselunterschied zwischen Tay-Sachs und Sandhoff-Krankheit ist, dass die Tay-Sachs-Krankheit eine lysosomale Speicherstörung ist, die auf den Mangel an β-Hexosaminidase A Enzym zurückzuführen ist, während die Sandhoff-Erkrankung eine lysosomale Speicherstörung aufgrund des Mangels von β-Hexosaminidase A und β-Hexosaminidase B Enzymiyme ist.
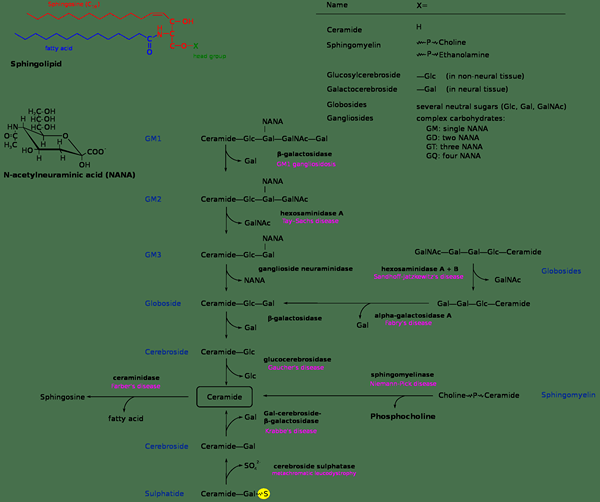
Ganglioside sind komplexe Sphingolipide im Gehirn. Es gibt zwei Hauptformen: GM1 und GM2. Beide Typen können an lysosomalen Speicherstörungen beteiligt sein. GM2 -Gangliosidosen sind eine Gruppe von drei verwandten rezentiv ererbten Krankheiten, bei denen ein Mangel an β -Hexosaminidase zur Akkumulation von GM2 -Gangliosid im Gehirn führt. Diese drei Krankheiten sind besser unter ihren individuellen Namen bekannt: Tay-Sachs-Krankheit, AB-Variante und Sandhoff-Krankheit.
INHALT
1. Überblick und wichtiger Unterschied
2. Was ist Tay-Sachs-Krankheit
3. Was ist Sandhoff -Krankheit
4. Ähnlichkeiten - Tay -Sachs und Sandhoff -Krankheit
5. Tay-Sachs gegen Sandhoff-Krankheit in tabellarischer Form
6. Zusammenfassung - Tay -Sachs gegen Sandhoff -Krankheit
Was ist Tay-Sachs-Krankheit?
Die Tay-Sachs-Krankheit ist eine lysosomale Speicherstörung, die aufgrund des Mangels des β-Hexosaminidase-A-Enzyms auftritt. Es handelt. Die häufigste Form ist die kindliche Tay-Sachs-Krankheit. Dies zeigt sich um drei bis sechs Monate, wenn Babys die Fähigkeit verlieren, sich umzudrehen, zu sitzen oder zu kriechen. Später folgt Anfälle, Hörverlust und Unfähigkeit, sich zu bewegen, wobei der Tod im Alter von drei bis fünf Jahren auftritt. Die weniger häufigen Formen sind die Tay-Sachs-Krankheit in späteren Kindheit oder im Erwachsenenalter (juvenile oder spät einsetzende). Diese Formen sind in der Regel weniger schwerwiegend. Die jugendliche Form führt jedoch normalerweise zum Tod bis zum Alter von 15 Jahren. Diese Krankheit hat eine ethnische Assoziation. Es ist selten in der allgemeinen Bevölkerung. Aber in aschkenasischen Juden, französischen Kanadiern im Südosten von Quebec, dem alten Orden Amish von Pennsylvania und Cajuns im südlichen Louisiana ist dieser Zustand häufiger.
Die Tay-Sachs-Krankheit wird durch genetische Mutation der verursacht Hexa Gen auf Chromosom 15. Dieses Gen kodiert für eine Untereinheit des β-Hexosaminidase-A-Enzyms. Die Mutation stört die Enzymaktivität, was zum Aufbau von GM2 -Gangliosid im Gehirn und im Rückenmark führt. Dies führt zu Toxizität. Dieser Zustand kann durch Messung des Blut-β-Hexosaminidase-A-Spiegels, der mikroskopischen Analyse des Netzhautneurons diagnostiziert werden. Darüber hinaus umfassen die möglichen Behandlungen für die Tay-Sachs-Erkrankung die Enzymersatztherapie, die Therapie der Substratreduktion, die Erhöhung der β-Hexosaminidase-A-Aktivität durch das Arzneimittelpyrimethamin, die Transplantation der Nabelschnurblut und die Gentherapie.
Was ist Sandhoff -Krankheit?
Sandhoff-Erkrankung ist eine lysosomale Speicherstörung. Es gibt ein kombiniertes β-.Hexosaminidase A- und B -Mangel an dieser Krankheit. Die klinische Manifestation dieser Krankheit umfasst die progressive Gehirndegeneration ab 6 Monaten, die von Blindheit, kirschrotem Makula und Hyperakusis begleitet wird. Es gibt drei Arten: die klassische kindliche Form, die im Alter von 2 bis 9 Monaten auftritt, die jugendliche Form, die bei Kindern im Alter von 3 bis 10 Jahren auftritt, und die Erwachsenenform, die bei älteren Erwachsenen auftritt. Die klassische kindliche Form verursacht den Tod bis zum 3. Lebensjahr, während die jugendliche Form den Tod bis zum Alter von 15 Jahren verursacht. Darüber hinaus ist noch nicht bekannt, ob diese Krankheit zu einer Verringerung der Lebensdauer älterer Erwachsener führt. Es gibt eine viszerale Beteiligung (Hepatomegalie und Knochenwandel) an dieser Krankheit. Es gibt jedoch keine ethnische Assoziation für Sandhoff -Erkrankungen
Die Diagnose dieser Krankheit kann durch Testen der Aktivität von β-Hexosaminidase A und B (Enzym-Assays) und deren Spiegel im Blut gestellt werden. Gentests der Hexb Gene kann auch verwendet werden, um die Diagnose zu bestätigen. Die anderen Diagnoseverfahren umfassen Leberbiopsie, molekulare Analyse von Zellen und Gewebe, um das Vorhandensein einer genetischen Stoffwechselstörung und Urinanalyse zu bestimmen. Darüber hinaus können die Behandlungsoptionen für Sandhoff-Erkrankungen die Verwendung von Antikonvulsiva zur Reduzierung von Anfällen, den Konsum einer präzisen Ernährung, der Unterstützung der Atemwege für Kinder, Medikamenten wie N-Butyl-Desoxynojirimycin und Gentherapie umfassen.
Was sind die Ähnlichkeiten zwischen Tay-Sachs und Sandhoff-Erkrankungen?
- Tay-Sachs und Sandhoff-Erkrankungen sind zwei Arten von GM2-Gangliosidosen.
- Bei beiden Krankheiten akkumulieren GM2 -Ganglioside in neuronalen Zellen.
- Beide Krankheiten sind auf den Mangel von zwei Arten von β-Hexosaminidase zurückzuführen
- Sie sind genetische Störungen, die in einem autosomalen rezessiven Muster vererbt wurden.
- Beide Krankheiten haben drei Formen: kindliche, jugendliche und erwachsene Einstellung.
- Sie zeigen ähnliche häufige Symptome.
- Darüber hinaus können sie durch Gentherapie behandelt werden.
Was ist der Unterschied zwischen Tay-Sachs und Sandhoff-Krankheit?
Die Tay-Sachs-Krankheit ist eine lysosomale Speicherstörung, die aufgrund des Mangels von β-Hexosaminidase-A-Enzym auftritt, während die Sandhoff-Erkrankung eine lysosomale Speicherstörung ist, die aufgrund des Mangels von β-Hexosaminidase A und β-Heaminidase B-Enzymmesse auftritt, und β-Hexosaminidase A und β-Heaminidase B-Enzymmes. Dies ist daher der wichtigste Unterschied zwischen Tay-Sachs und Sandhoff-Erkrankungen. Darüber hinaus ist die Tay-Sachs-Erkrankung auf die genetische Mutation des Hexa Gene, während Sandhoff -Erkrankung auf die genetische Mutation der zurückzuführen ist Hexb Gen.
Die folgende Infografik zeigt die Unterschiede zwischen Tay-Sachs und Sandhoff-Erkrankungen in tabellarischer Form für Seite an Seitenvergleich.
Zusammenfassung -Tay -Sachs gegen Sandhoff -Krankheit
Tay-Sachs und Sandhoff-Erkrankungen sind zwei Arten von lysosomalen Speicherstörungen, die aufgrund der Ansammlung von GM2-Gangliosid auftreten. Die Tay-Sachs-Erkrankung ist auf den Mangel an β-Hexosaminidase-A-Enzym zurück. Dies ist also der Hauptunterschied zwischen Tay-Sachs und Sandhoff-Krankheit.
Referenz:
1. "Tay-Sachs-Krankheit.”Healthline, Healthline Media, 3. März. 2022.
2. „Sandhoff -Krankheit: Was ist es, Ursachen, Diagnose und Behandlung.Cleveland Clinic.
Bild mit freundlicher Genehmigung:
1. "Sphingolipidosen" von eBuxbaum, Sav_vas - eigene Arbeit (CC BY -SA 3.0) über Commons Wikimedia