Unterschied zwischen Alpha und Beta Thalassämie
Schlüsselunterschied - Alpha vs Beta Thalassämie
Thalassämie ist eine heterogene Gruppe von Erkrankungen, die durch ererbte Mutationen verursacht werden. Es gibt zwei Hauptformen von Thalassämie wie Alpha Thalassämie und Beta Thalassämie. Bei Alpha-Thalassämie ist die Anzahl der Alpha-Globin-Ketten abgenommen, während es bei Beta-Thalassämie die Anzahl der Beta-Globin-Ketten ist, die untergehen. Dies ist der Hauptunterschied zwischen Alpha und Beta Thalassämie.
INHALT
1. Überblick und wichtiger Unterschied
2. Was Alpha Thalassämie
3. Was für eine Beta -Thalassämie
4. Ähnlichkeiten zwischen Alpha und Beta Thalassämie
5. Seite an Seitenvergleich - Alpha gegen Beta -Thalassämie in tabellarischer Form
6. Zusammenfassung
Was ist Alpha Thalassämie?
In Alpha Thalassämie werden einige der Gene, die für die Kodierung der Alpha -Globin -Ketten verantwortlich sind, gelöscht. Das Alpha -Globin -Gen hat im Allgemeinen vier Kopien. Die Schwere der Krankheit hängt von der Anzahl fehlender Kopien ab.
Hydrops Fetalis
Die Synthese von Alpha -Globin -Ketten ist vollständig unterdrückt, wenn alle vier Kopien des Alpha -Globin -Gens fehlen. Da Alpha -Globin -Ketten für die Synthese sowohl des fetalen als auch des adulten Hämoglobins benötigt werden, ist dieser Zustand nicht mit dem Leben kompatibel. Daher tritt bei der Utero -Beendigung der Schwangerschaft auf, wenn der Fötus durch diesen Zustand beeinflusst wird.
HBH -Krankheit
Dieser Zustand wird durch das Fehlen von drei Kopien des Alpha -Globin -Gens verursacht. Dies führt zu einer mittelschweren bis schweren hypochromischen mikrozytischen Anämie mit assoziiertem Splenomegalie.
Alpha -Thalassämie -Eigenschaften
Dies ist durch die Abwesenheit oder die Inaktivität von einem oder zwei Kopien des Alpha -Globin -Gens verursacht. Obwohl die Alpha -Thalassämie -Merkmale keine Anämie verursachen, können sie das mittlere korpuskuläre Volumen und die mittleren korpuskulären Hämoglobinspiegel unterziehen, während die Anzahl der roten Blutkörperchen über 5 erhöht wird.5*1012/L.
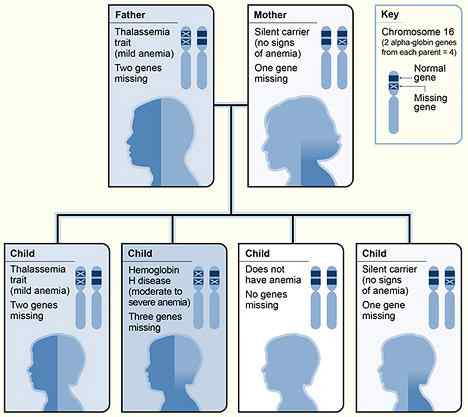
Abbildung 01: Vererbung von Alpha Thalassämie
Die Diagnose von Alpha -Thalassämie erfolgt durch die Globin -Kettensynthesestudien.
Management
Patienten mit einer milden Form von Anämie benötigen normalerweise keine Behandlungen. Die Verabreichung von Eisen und Folsäure wird nur bei einigen Patienten befürwortet. Diejenigen mit den schweren Formen von Alpha -Thalassämie benötigen eine lebenslange Bluttransfusion.
Was ist Beta Thalassämie?
Bei Beta -Thalassämie sinkt die Anzahl der Beta -Globin -Ketten aus.
Beta Thalassämie Major
Wenn beide Elternteile der Beta -Thalassämie -Eigenschaft sind, beträgt die Möglichkeit eines Nachkommens, der Beta Thalassämie -Hauptfach erbt. In Beta Thalassämie -Hauptfach wird die Produktion von Beta -Globin -Ketten entweder vollständig unterdrückt oder drastisch reduziert. Da es nicht genügend Beta -Globin -Ketten gibt, mit denen sie sich kombinieren können. Dies führt zu einer vorzeitigen Hämolyse von roten Zellen und einer ineffektiven Erythropoese.
Klinische Merkmale
- Schwere Anämie, die nach 3 bis 6 Monaten nach der Geburt offensichtlich wird.
- Splenomegalie und Hepatomegalie
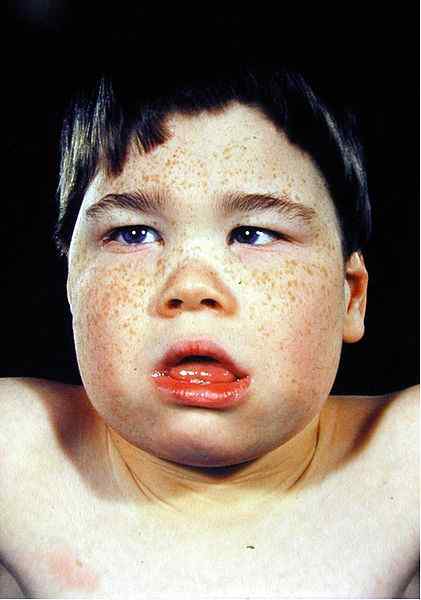
- Thalassämiefazies
Die Änderungen der Gesichtsmerkmale sind auf die Ausdehnung von Knochen aufgrund von Knochenmarkhyperplasie zurückzuführen. Das Röntgenröntgenaufbau zeigt das Aussehen des Schädels, der typischerweise bei Beta-Thalassämie zu sehen ist.
Abbildung 02: Thalassämiefazies
Labor Diagnose
Hochleistungsflüssigchromatographie (HPLC) ist die Hauptmethode zur Diagnose von hämatologischen Erkrankungen heutzutage. Beta -Thalassämie -Haupt -HPLC zeigt das Vorhandensein von reduzierten HBA -Werten mit ungewöhnlich hohen HBF -Werten. Eine volle Blutzahl wird die Existenz einer hypochromischen mikrozytischen Anämie zeigen, und die Untersuchung eines Blutfilm.
Behandlung
- Regelmäßige Bluttransfusionen
- Eisen -Chelationstherapie
- Folsäure (wenn die Nahrungsaufnahme von Folsäure nicht zufriedenstellend ist)
- Splenektomie (manchmal verwendet, um den Blutbedarf zu verringern)
- Knochenmarktransplantation
- Gentherapie zum Screening und therapeutischen Zwecken
Beta Thalassämie Merkmal/Minderjähriger
Beta -Thalassämie -Moll ist eine häufige Erkrankung, die häufig symptomlos ist. Obwohl die Anzeichen und Symptome denen von Alpha Thalassämie ähnlich sind, ist Beta Thalassämie schwerwiegender als sein Gegenstück. Die Diagnose von Beta -Thalassämie -Moll wird gestellt, wenn die HBA2 Level beträgt mehr als 3.5%.
Thalassämie intermedia
Thalassämie intermedia bezieht.
Was ist die Ähnlichkeit zwischen Alpha und Beta Thalassämie?
- Unter beiden Bedingungen kommt es zu einer Abnahme des Hämoglobinspiegels von Blut.
Was ist der Unterschied zwischen Alpha und Beta Thalassämie?
Alpha gegen Beta Thalassämie | |
| Die Anzahl der Alpha -Globin -Ketten verringert sich. | Die Anzahl der Beta -Globin -Ketten verringert sich. |
| Löschung von Genen | |
| Einige der für die Kodierung der Alpha -Globinketten verantwortlichen Gene werden gelöscht. | Gene, die für die Synthese von Beta -Globin -Ketten verantwortlich sind, sind entweder teilweise oder vollständig gelöscht. |
| Typen | |
| Hydrops Fetalis, HBH -Krankheit und Alpha -Thalassämie -Merkmale sind die Hauptformen von Alpha -Thalassämie. | Es gibt zwei Hauptformen von Beta Thalassämie als Beta -Thalassämie Major und Beta Thalassämie -Moll. |
| Diagnose | |
| Die Diagnose von Alpha -Thalassämie erfolgt durch die Globin -Kettensynthesestudien. | Hochleistungs-Flüssigchromatographie (HPLC) ist die Untersuchung zur Diagnose einer Beta-Thalassämie. |
| Klinische Merkmale | |
|
|
| Behandlung und Management | |
|
|
Zusammenfassung -Alpha gegen Beta -Thalassämie
Thalassämie ist eine heterogene Gruppe von Störungen, die durch ererbte Mutationen verursacht werden, die die Synthese von Alpha- oder Beta -Globin -Ketten verringern, aus denen das adulte Hämoglobin HBA besteht. Thalassämie kann weitgehend in zwei Hauptkategorien eingeteilt werden wie Alpha Thalassämie und Beta Thalassämie. Bei Alpha-Thalassämie wird die Menge an Alpha-Ketten verringert, und bei Beta-Thalassämie wird die Anzahl der Beta-Ketten verringert. Dies ist der Hauptunterschied zwischen Alpha und Beta Thalassämie.
Laden Sie die PDF -Version von Alpha vs Beta Thalassämie herunter
Sie können die PDF -Version dieses Artikels herunterladen und ihn für Offline -Zwecke gemäß Citation Note verwenden. Bitte laden Sie die PDF -Version hier den Unterschied zwischen Alpha und Beta Thalassämie herunter
Referenz:
1.Kumar, Parveen J., und Michael l. Clark. Kumar & Clark Klinische Medizin. Edinburgh: w.B. Saunders, 2009.
Bild mit freundlicher Genehmigung:
1. "Thalassämie Alpha" von National Heart Lung and Blood Institute (NIH) - National Heart Lung and Blood Institute (NIH) (Öffentlichkeit) über Commons Wikimedia
2. "ATR-X" von Gibbons r. - Gibbons r. Alpha Thalassämie-Mental-Retardation, x verknüpft. Orphanet J seltene dis. 1, 15. 2006. doi: 10.1186/1750-1172-1-15. PMID 16722615 (CC von 2.0) über Commons Wikimedia