Unterschied zwischen Sichelzellenanämie und Thalassämie
Schlüsselunterschied - Sichelzellenanämie gegen Thalassämie
Thalassämie ist eine heterogene Gruppe von Störungen, die durch ererbte Mutationen verursacht werden. Sichelzellenanämie ist eine schwere erbliche Form von Anämie, bei der eine mutierte Form von Hämoglobin die roten Blutkörperchen in einer Halbmondform bei niedrigen Sauerstoffspiegeln verzerrt. Der Hauptunterschied zwischen Sichelzellenanämie und Thalassämie ist das in Thalassämie beide α Und β Globinketten können betroffen sein, aber bei Sichelzellenanämie nur die β Globinketten sind betroffen.
INHALT
1. Überblick und wichtiger Unterschied
2. Was ist Thalassämie
3. Was ist Sichelzellenanämie
4. Ähnlichkeiten zwischen Sichelzellenanämie und Thalassämie
5. Seite an Seitenvergleich - Sichelzellenanämie gegen Thalassämie in tabellarischer Form
6. Zusammenfassung
Was ist Thalassämie?
Thalassämie ist „eine heterogene Gruppe von Erkrankungen, die durch ererbte Mutationen verursacht werden, die die Synthese von Alpha- oder Beta -Globinketten verringern, die das adulte Hämoglobin -HBA bestehen, das zur Anämie, der Gewebehypoxie und der roten Zell -Hämolyse im Zusammenhang mit dem Ungleichgewicht der Globinkettensynthese im Zusammenhang mit der Synthese der Globinketten -Kettensynthese führt.“.
Thalassämie kann weitgehend in zwei Hauptkategorien eingeteilt werden.
- Alpha Thalassämie
- Beta Thalassämie.
Alpha Thalassämie
In Alpha Thalassämie werden einige der für die Kodierung der Alpha -Globin -Ketten verantwortlichen Gene gelöscht. Normalerweise hat das Alpha Globin -Gen vier Kopien. Die Schwere der Krankheit hängt von der Anzahl solcher Kopien ab, die fehlen.
1. Hydrops Fetalis
Wenn alle vier Kopien des Alpha -Globin -Gens fehlen, wird die Synthese von Alpha -Globin -Ketten vollständig unterdrückt. Da Alpha -Globin -Ketten für die Synthese sowohl des fetalen als auch des adulten Hämoglobins benötigt werden, ist diese Erkrankung nicht mit dem Leben kompatibel und führt daher zum Tod in der Gebärmutter.
2. HBH -Krankheit
Das Fehlen von drei Kopien des Alpha -Globin -Gens führt zu einer mittelschweren bis schweren hypochromischen mikrokytischen Anämie mit assoziiertem Splenomegalie.
3. Alpha -Thalassämie -Eigenschaften
Diese sind auf die Abwesenheit oder die Inaktivität von einem oder zwei Kopien des Alpha -Globin -Gens zurückzuführen. Obwohl die Alpha -Thalassämie -Merkmale keine Anämie verursachen, können sie das mittlere korpuskuläre Volumen und die mittleren korpuskulären Hämoglobinspiegel verringern, während die Anzahl der roten Blutkörperchen über 5 erhöht wird.5*1012/L.
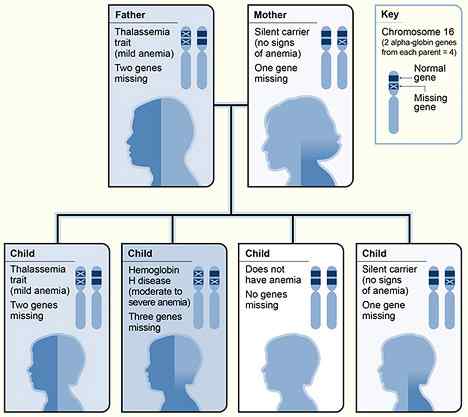
Abbildung 01: Vererbung von Alpha Thalassämie
Beta -Thalassämie -Syndrome
1. Beta Thalassämie Major
Wenn beide Eltern die Fluggesellschaften der Beta -Thalassämie -Eigenschaft sind, beträgt die Chance, dass ein Nachwuchs die Beta -Thalassämie -Hauptfach erhält, 25%. In diesem Zustand wird die Produktion von Beta -Globin -Ketten entweder vollständig unterdrückt oder drastisch reduziert. Da es nicht genügend Beta -Globin -Ketten gibt, mit denen sie sich kombinieren können. Dies ebnet den Weg für eine vorzeitige Hämolyse von roten Zellen und ineffektiven Erythropoese.
Klinische Merkmale
- Schwere Anämie, die nach 3 bis 6 Monaten nach der Geburt offensichtlich wird.
- Splenomegalie und Hepatomegalie
- Thalassämiefazies
Labor Diagnose
Die Haupttechnik zur Diagnose hämatologischer Erkrankungen heutzutage ist die Hochleistungs-Flüssigchromatographie (HPLC). In Beta Thalassämie-Hauptfach zeigt HPLC das Vorhandensein vielreduzierter HBA-Werte mit ungewöhnlich hohen HBF-Werten. Eine vollständige Blutzahl wird die Existenz einer hypochromischen mikrokytischen Anämie zeigen, während die Untersuchung eines Blutfilm.
Behandlung
- Regelmäßige Bluttransfusionen
- Folsäure wird angegeben, wenn die Nahrungsaufnahme von Folsäure nicht zufriedenstellend ist.
- Eisen -Chelationstherapie
- Manchmal wird auch Splenektomie durchgeführt, um den Blutbedarf zu verringern.
2. Beta Thalassämie Merkmal/Minderjähriger
Beta -Thalassämie -Moll ist eine häufige Erkrankung, die die meiste Zeit symptomlos ist. Obwohl die Merkmale denen von Alpha Thalassämie ähnlich sind, ist die Beta -Thalassämie schwerwiegender als sein Gegenstück. Die Diagnose von Beta -Thalassämie -Moll wird gestellt, wenn die HBA2 Level beträgt mehr als 3.5%.
3. Thalassämie intermedia
Fälle von Thalassämie mäßiger Schwere, die keine regelmäßigen Transfusionen benötigen, werden als Thalassämie intermedia bezeichnet.
Was ist Sichelzellenanämie?
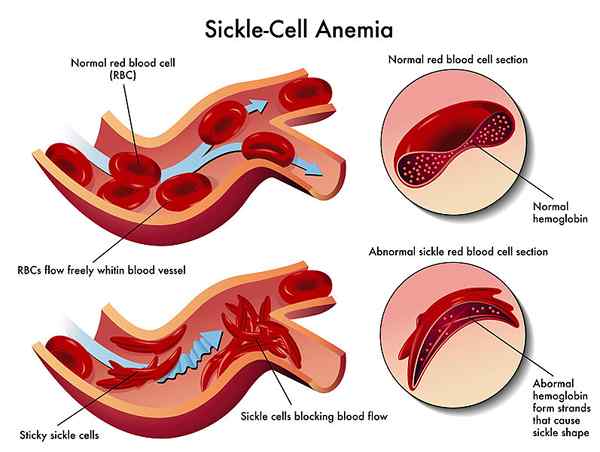
Sichelzellenanämie ist eine schwere erbliche Form von Anämie, bei der eine mutierte Form von Hämoglobin die roten Blutkörperchen in einer Halbmondform bei niedrigen Sauerstoffspiegeln verzerrt.
Die Form von Hämoglobin erzeugt aufgrund der genetischen Mutation bei homozygoter Sichelzellenanämie als HBSS (Sichel -Hämoglobin) bezeichnet. Wenn der Sauerstoffspiegel unter einen bestimmten kritischen Punkt fällt, bildet Sichel-Hämoglobin durch einen Polymerisationsprozess Kristalle. Es ist diese Polymerisation, die die roten Blutkörperchen in eine Halbmondform verzerrt. Da die verzerrten roten Zellen ihre Einhaltung verloren haben, können sie nicht mehr durch winzige Blutgefäße gelangen. In diesen winzigen Gefäßen blockieren Sichelzellen das Lumen und beeinträchtigen die Blutversorgung der Organe, die zu mehreren Infarkten führen.
Abbildung 02: Sichelzellenanämie
Klinische Merkmale der Sichelzellenanämie
Eine durch Krisen unterbrochene schwere hämolytische Anämie kann beobachtet werden.
1. Vaso okklusive Krisen
Faktoren wie Infektionen, Dehydratisierung, Azidose und Desoxygenierung, die für die vaso okklusiven Krisen prädisponiert werden. Der Patient beschwert sich über starke Schmerzen in den Extremitäten. Dies liegt an den Infarkten in den kleinen Knochen der Gliedmaßen.
2. Viszerale Sequestrierungskrisen
Diese sind an der Bündelung von Blut und Klingen, die in den Organen stattfinden. Die tödlichste Komplikation dieser Art ist das Sichel -Brust -Syndrom, bei dem der Patient mit Dyspnoe und Brustschmerzen auftritt. Lungeninfiltrate sind auf der Röntgenaufnahme der Brust zu sehen.
3. Aplastische Krisen
Aplastische Krisen sind durch den plötzlichen Abfall des Hämoglobinspiegels nach einer Parvo -Virus -Infektion gekennzeichnet. Folatmangel kann auch einen Beitragsfaktor sein.
Andere klinische Merkmale der Sichelzellenanämie
- Geschwüre in den unteren Gliedern
- Pulmonale Hypertonie
- Gallensteine
Labordiagnose einer Sichelzellenanämie
- Der Hämoglobinspiegel beträgt normalerweise 6-9g/dl.
- Vorhandensein von Sichelzellen und Zielzellen im Blutfilm.
- Screening -Tests auf Sauberling mit Chemikalien wie Dithionat sind positiv, wenn das Blut deoxygeniert ist.
- In HPLC wird HBA nicht erkannt.
Behandlung von Sichelzellenanämie
- Vermeiden Sie die Faktoren, von denen bekannt ist, dass sie die Krisen auslösen.
- Folsäure.
- Gute Ernährung und Hygiene.
- Pneumokokken-, Haemophilus- und Meningokokkenimpfung.
- Krisen sollten gemäß der Erkrankung, des Alters und der Einhaltung des Arzneimittels des Patienten behandelt werden.
Was sind die Ähnlichkeiten zwischen Sichelzellenanämie und Thalassämie?
- Sowohl Thalassämie als auch Sichelzellenanämie sind genetische Erkrankungen, die die Struktur und Funktion von Hämoglobin beeinflussen.
Was ist der Unterschied zwischen Sichelzellenanämie und Thalassämie?
Sichelzellenanämie gegen Thalassämie | |
| Sichelzellenanämie ist eine schwere erbliche Form von Anämie, bei der eine mutierte Form von Hämoglobin die roten Blutkörperchen in einer Halbmondform bei niedrigen Sauerstoffspiegeln verzerrt. | Thalassämie ist eine heterogene Gruppe von Störungen, die durch ererbte Mutationen verursacht werden. |
| Art der betroffenen Globinkette | |
| In Sichelzellenanämie sind nur Beta -Ketten betroffen. | Sowohl Alpha- als auch Beta -Ketten können bei Thalassämie betroffen sein. |
| Art der genetischen Mutation | |
| Der genetische Defekt, der Sichelzellenanämie verursacht. | Thalassämie wird entweder durch eine Punktmutation oder durch eine Gendeletion verursacht. |
| Widerstand gegen Malaria | |
| Es ist bekannt, dass der genetische Defekt, der Sichelzellenanämie verursacht. | Genetischer Defekt bei Thalassämie liefert keine Resistenz gegen Malaria. |
| Genetischer Ursprung | |
| Sichelzellenanämie ist eine autosomale rezessive Störung. | Thalassämie ist eine autosomale kodominante Störung. |
Zusammenfassung -Sichelzellenanämie gegen Thalassämie
Thalassämie und Sichelzellenanämie sind zwei schwerwiegende hämatologische Störungen, die hauptsächlich in der pädiatrischen Praxis auftreten. Daher ist es wichtig, den Unterschied zwischen Sichelzellenanämie und Thalassämie klar zu verstehen. Das Erhöhen des Gemeinschaftsbewusstseins für diese Krankheiten wird hilfreich sein, um die Inzidenz von ihnen zu minimieren, da die Konsangerheime eine wichtige Rolle bei der Übertragung dieser genetischen Störungen von Generation zu Generation spielen.
Laden Sie die PDF -Version von Sichelzellenanämie gegen Thalassämie herunter
Sie können die PDF -Version dieses Artikels herunterladen und ihn für Offline -Zwecke gemäß Zitatnotizen verwenden. Bitte laden Sie die PDF -Version hier den Unterschied zwischen Sichelzellenanämie und Thalassämie herunter.
Verweise:
1. Hoffbrand, a. V., und P. A. H. Moos. Wesentliche Hämatologie. 6. ed. Oxford: Wiley-Blackwell, 2011. Drucken.
2. Kumar, Vinay, Stanley Leonard Robbins, Ramzi S. Cotran, Abul K. Abbas und Nelson Fausto. Robbins und Cotran pathologische Grundlage der Krankheit. Philadelphia, PA: Elsevier Saunders, 2010. Drucken.
Bild mit freundlicher Genehmigung:
1. "Thalassämie Alpha" von National Heart Lung and Blood Institute (NIH) - National Heart Lung and Blood Institute (NIH) (Öffentlichkeit) über Commons Wikimedia
2. "Risiko-Faktoren-für-Slipple-Cell-Anämie (1) 2" von Diana Grib-eigene Arbeit (CC BY-SA 4.0) über Commons Wikimedia