Unterschied zwischen Sichelzellenerkrankungen und Sichelzellenanämie
Schlüsselunterschied - Sichelzellenerkrankung gegen Sichelzellenanämie
Sichelzellenerkrankung ist eine häufige erbliche Hämoglobinopathie, die durch eine Punktmutation in Beta -Globin verursacht wird, die die Polymerisation von deoxygeniertem Hämoglobin fördert, was zu einer Verzerrung der roten Zellen, einer hämolytischen Anämie, der Obstruktion der mikrischen Gefäßverstopfung und einer ischämischen Gewebeschäden führt. Sichelzellenanämie ist eine schwere erbliche Anämieform, die sich aus der Sichelzellenerkrankung ergibt. Sichelzellenerkrankung hat eine Gruppe pathologischer Manifestationen während Sichelzellenanämie ist eine solche pathologische Manifestation der Sichelzellenerkrankung. Dies ist der wichtigste Unterschied zwischen Sichelzellenerkrankungen und Sichelzellenanämie.
INHALT
1. Überblick und wichtiger Unterschied
2. Was ist Sichelzellenerkrankung
3. Was ist Sichelzellenanämie
4. Ähnlichkeiten zwischen Sichelzellenerkrankungen und Sichelzellenanämie
4. Seite an Seitenvergleich - Sichelzellenerkrankung gegen Sichelzellenanämie in tabellarischer Form
5. Zusammenfassung
Was ist Sichelzellenerkrankung?
Sichelzellenerkrankung ist eine häufige erbliche Hämoglobinopathie, die durch eine Punktmutation in Beta -Globin verursacht wird, die die Polymerisation von deoxygeniertem Hämoglobin fördert.
Hämoglobin hat eine tetramere Struktur, die sich aus zwei Paar Alpha- und Beta -Ketten zusammensetzt. Normale adulte rote Zellen haben HBA (α)2 β2) als ihre dominante Form von Hämoglobin. Bei Sichelzellenerkrankung wird der Glutamatrest im sechsten Codon des Beta -Globin -Gens durch Valin ersetzt. Diese Substitution führt zu verschiedenen strukturellen und funktionellen Veränderungen im Hämoglobinmolekül. Neben HBA haben Menschen, die an der Sichelzellenerkrankung leiden, eine spezielle Art von Hämoglobin in ihren roten Zellen, die als Sichel -Hämoglobin (HBS) bezeichnet werden.
Pathogenese der Sichelzellenerkrankung
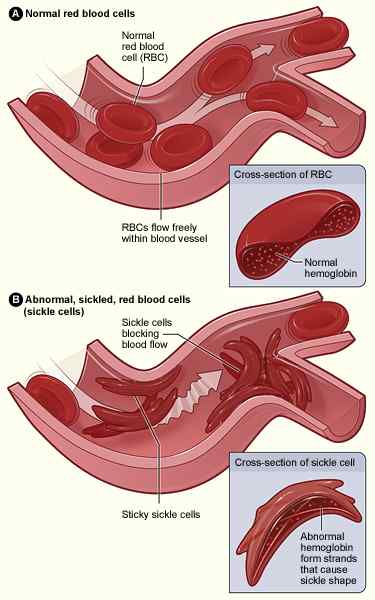
Frei fließendes Cytosol der roten Blutkörperchen verwandelt sich in ein viskoses Gel, wenn der Teildruck von Sauerstoff unter ein bestimmtes kritisches Niveau fällt. Mit fortgesetzter Desoxygenierung polymerisieren HBS -Moleküle in lange Fasern in den roten Zellen, die sie in eine Halbmondform verzerren. Dies ist die pathologische Grundlage für die Hauptmanifestationen wie chronische Hämolyse, Mikrogefäßerverschluss und Gewebeschäden.
Wenn die HBS -Polymere wachsen. Diese strukturelle Modifikation der roten Blutkörperchen induziert einen Zustrom von CA2+.Erhöhter intrazellulärer Calciumspiegel induziert dann die Kreuzverbindung der intrazellulären Proteine, was zu einem Ausfluss von k führt+ und Wasser. Die Wiederholung dieses Prozesses entwässert die roten Blutkörperchen und macht sie starr und dicht. Letztendlich werden sie irreversibel krankende Zellen, die durch extravaskuläre Hämolyse schnell aus der Kreislauf entfernt werden.
Es gibt mehrere Vorstellungen über die pathologische Grundlage der Mikrogefäßerverschluss, aber der genaue Mechanismus ist nicht klar verstanden.
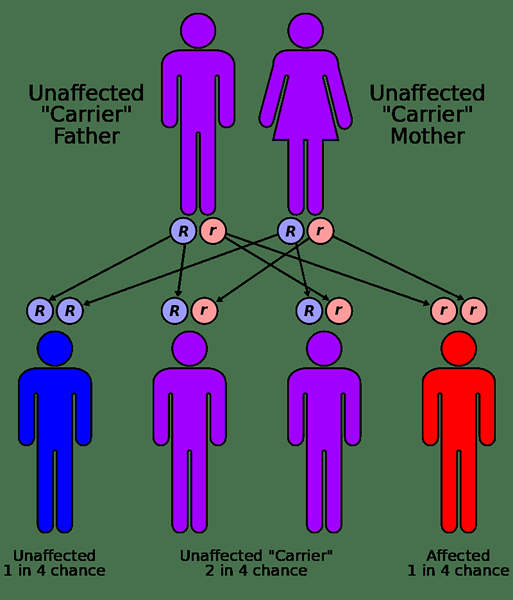
Abbildung 01: Sichelzellenerkrankung wird im autosomalen rezessiven Muster vererbt.
Klinische Merkmale von Sichelzellenerkrankungen
Sichelzellenerkrankung hat ein breites Spektrum klinischer Manifestationen. Einige der betroffenen Personen können verkrüppelt werden, während andere nur leichte Symptome haben können.
(Sowohl Sichelzellenerkrankungen als auch Sichelzellenanämie haben viele häufige klinische Manifestationen, die unter der Überschrift „klinische Merkmale der Sichelzellenanämie“ diskutiert werden))
Diagnose einer Sichelzellenerkrankung
- Hämoglobinelektrophorese, um das Vorhandensein von HBs zu demonstrieren
- Dithionat -Test
- Die pränatale Diagnose ist durch die Analyse der durch Amniozentese erhaltenen fetalen DNA möglich.
Was ist Sichelzellenanämie?
Die schwere erbliche Form der Anämie, die sich infolge der Sichelzellenerkrankung ergibt.
(Pathogenese der Sichelzellenanämie wurde unter der Überschrift „Pathogenese der Sichelzellenerkrankung“ diskutiert.)
Abbildung 02: Sichelzellen
Klinische Merkmale der Sichelzellenanämie
Klinische Merkmale sind eine schwere hämolytische Anämie, die von Krisen unterbrochen wird. Es kann vier Haupttypen von Krisen geben.
1. Vaso okklusive Krisen
Vaso okklusive Krisen sind häufiger als die anderen und werden durch Faktoren wie Infektionen, Azidose, Dehydration und Desoxygenierung prädisponiert. Aufgrund der Blockierung der Blutgefäße wird die Blutversorgung bestimmter Körperbereiche, insbesondere an den Extremitäten, beeinträchtigt. Infolgedessen treten in diesen Regionen Infarkte auf, was zu intensiven Schmerzen führt. Es gibt eine Erkrankung, die als Hand -Fuß -Syndrom bezeichnet wird, bei der der Patient über starke Schmerzen in den Extremitäten klagt. Dies geschieht aufgrund der Infarkte in den kleinen Knochen der Gliedmaßen.
2. Viszerale Sequestrierungskrisen
Diese Krisen treten infolge des Schalllings und des Blutverpackens in den Organen auf. Anämie verschärft sich während einer viszeralen Sequestrierungskrise auf ein schwerwiegendes Niveau. Das akute Brustsyndrom ist die gefährlichste Komplikation dieser Krise. Patienten leiden unter Schmerzen in der Brust und einer Dyspnoe. Eine Röntgenaufnahme der Brust zeigt das Vorhandensein von Lungeninfiltraten.
3. Aplastische Krisen
Diese treten nach einer Parvo -Virusinfektion auf und manchmal auch wegen Folatmangel. Aplastische Krisen sind durch den plötzlichen Tropfen des Hämoglobinspiegels gekennzeichnet, der häufig Transfusion erfordert.
4. Hämolytische Anämie
Andere klinische Merkmale der Sichelzellenanämie
- Geschwüre im unteren Glied.
- Milz wird im Säuglingsalter vergrößert, wird jedoch aufgrund von Infarkten allmählich verringert (Autosplenktomie).
- Pulmonale Hypertonie.
Labordiagnose einer Sichelzellenanämie
- Der Hämoglobinspiegel beträgt normalerweise 6-9g/dl.
- Vorhandensein von Sichelzellen und Zielzellen im Blutfilm.
- Screening -Tests auf Sauberling mit Chemikalien wie Dithionat sind positiv, wenn das Blut deoxygeniert ist.
- In HPLC wird HBSS als dominante Form von Hämoglobin festgestellt und HBA wird nicht erkannt.
Behandlung für Sichelzellenanämie
- Vermeiden Sie die Faktoren, von denen bekannt ist, dass sie die Krisen auslösen.
- Folsäure.
- Gute Ernährung und Hygiene.
- Pneumokokken, Haemophilus und Meningokokkenimpfung.
- Krisen sollten gemäß der Erkrankung, des Alters und der Einhaltung des Arzneimittels des Patienten behandelt werden.
Was sind die Ähnlichkeiten zwischen Sichelzellenerkrankungen und Sichelzellenanämie?
- Sowohl Sichelzellenerkrankungen als auch Sichelzellenanämie werden durch dieselbe genetische Mutation verursacht, die die Beta -Globin -Ketten und damit die Struktur und Funktion von Hämoglobin beeinflusst.
- Da Sichelzellenanämie eine pathologische Manifestation der Sichelzellenerkrankung ist, teilen sie auch gemeinsame klinische Merkmale.
Was ist der Unterschied zwischen Sichelzellenerkrankungen und Sichelzellenanämie?
Sichelzellenkrankheit gegen Sichelzellenanämie | |
| Sichelzellenerkrankung ist eine häufige erbliche Hämoglobinopathie, die durch eine Punktmutation in Beta-Globin verursacht wird, die die Polymerisation von deoxygeniertem Hämoglobin fördert, was zu einer Verzerrung der roten Zellen, einer hämolytischen Anämie, der Schädigung der mikrogefäschelförmigen Obstruktion und einer Schädigung des ischämischen Gewebes führt | Sichelzellenanämie ist eine schwere erbliche Anämieform, die sich aus der Sichelzellenerkrankung ergibt. |
| Pathologische Manifestationen | |
| Sichelzellenerkrankung hat mehrere pathologische Manifestationen. | Sichelzellenanämie ist eine solche pathologische Manifestation der Sichelzellenerkrankung. |
Zusammenfassung -Sichelzellenerkrankung gegen Sichelzellenanämie
Sowohl Sichelzellenerkrankungen als auch Sichelzellenanämie sind häufige erbliche Erkrankungen, und angemessene Behandlungen können hilfreich sein, um den Lebensstandard des Patienten zu verbessern. Sichelzellenerkrankung hat eine Gruppe pathologischer Manifestationen, während Sichelzellenanämie eine solche pathologische Manifestation der Sichelzellenerkrankung ist. Dies ist der Hauptunterschied zwischen Sichelzellenerkrankungen und Sichelzellenanämie.
Laden Sie die PDF -Version von Sichelzellenkrankheiten gegen Sichelzellenanämie herunter
Sie können die PDF -Version dieses Artikels herunterladen und ihn für Offline -Zwecke gemäß Zitatnotizen verwenden. Bitte laden Sie die PDF -Version hier den Unterschied zwischen Sichelzellenerkrankungen und Sichelzellenanämie herunter.
Verweise:
1. Hoffbrand, a. V., und P. A. H. Moos. Wesentliche Hämatologie. 6. ed. Oxford: Wiley-Blackwell, 2011. Drucken.
2. Kumar, Vinay, Stanley Leonard Robbins, Ramzi S. Cotran, Abul K. Abbas und Nelson Fausto. Robbins und Cotran pathologische Grundlage der Krankheit. Philadelphia, PA: Elsevier Saunders, 2010. Drucken.
Bild mit freundlicher Genehmigung:
1. "Autorness“ von EN: Benutzer: Cburnett - eigene Arbeit in Inkscape, CC BY -SA 3.0) über Commons Wikimedia
2. "Sichelzelle 01" von National Heart, Lung und Blood Institute (NHLBI) - (Public Domain) über Commons Wikimedia