Was ist der Unterschied zwischen Phenylketonurie und Galactosämie
Der Schlüsselunterschied zwischen Phenylketonurie und Galaktosämie ist, dass Phenylketonurie aufgrund der Ansammlung einer Aminosäure verursacht wird, die Phenylalanin in verschiedenen Körperorganen namens.
Phenylketonurie und Galaktosämie sind zwei ererbte Stoffwechselstörungen. Erbliche Stoffwechselstörungen beziehen sich auf verschiedene Arten von Erkrankungen, die durch genetische Defekte oder Mutationen verursacht werden. Am häufigsten werden sie sowohl von den Eltern geerbt und stören den Stoffwechsel des Körpers. Diese Bedingungen können auch als angeborene Fehler des Stoffwechsels bezeichnet werden.
INHALT
1. Überblick und wichtiger Unterschied
2. Was ist Phenylketonurie
3. Was ist Galactosämie
4. Ähnlichkeiten - Phenylketonurie und Galaktosämie
5. Phenylketonurie gegen Galactosämie in tabellarischer Form
6. Zusammenfassung - Phenylketonurie gegen Galactosämie
Was ist Phenylketonurie?
Phenylketonurie ist eine seltene erbte Stoffwechselstörung. Es tritt aufgrund der Ansammlung einer Aminosäure auf, die Phenylalanin in verschiedenen Organen des Körpers bezeichnet hat. Phenylketonurie wird durch einen Defekt im Gen verursacht (PAH) Das kodiert das Enzym, das zum Abbau von Phenylalanin erforderlich ist. Ohne dieses Enzym tritt ein gefährlicher Aufbau dieser Aminosäure auf, wenn die Person Lebensmittel isst, die Eiweiß oder Aspartam isst (ein offizieller Süßstoff). Dies führt schließlich zu schwerwiegenden medizinischen Problemen. Für den Rest ihres Lebens müssen Menschen mit PKU (Babys, Kinder und Erwachsenen) einer Diät folgen, die Phenylalanin einschränkt. Darüber hinaus werden Babys in den USA und viele andere Länder kurz nach der Geburt auf PKU untersucht. Das Erkennen von PKU auf die richtige Weise kann dazu beitragen, Gesundheitsprobleme zu verhindern.
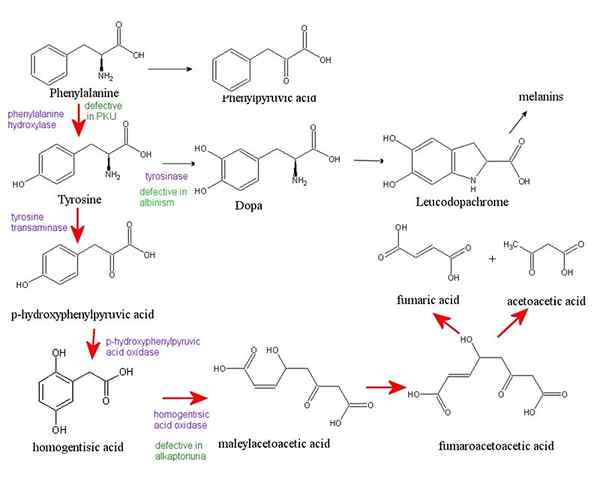
Abbildung 01: Phenylketonurie
PKU -Anzeichen und -Symptome können mild oder schwerwiegend sein, und sie umfassen einen muffigen Geruch im Atem, neurologische Probleme wie Anfälle, Hautausschläge, helle Haut und blaue Augen, ungewöhnlich kleiner Kopf, Hyperaktivität, intellektuelle Behinderung, verzögerte Entwicklung, Verhalten, emotional und soziale Probleme und psychiatrische Störungen. Dieser Zustand kann durch Familienanamnese, Blutuntersuchungen und Gentests diagnostiziert werden. Darüber hinaus sind eine lebenslange Ernährung mit einer sehr begrenzten Aufnahme von Protein-, neutraler Aminosäuretherapie und PKU.
Was ist Galactosämie?
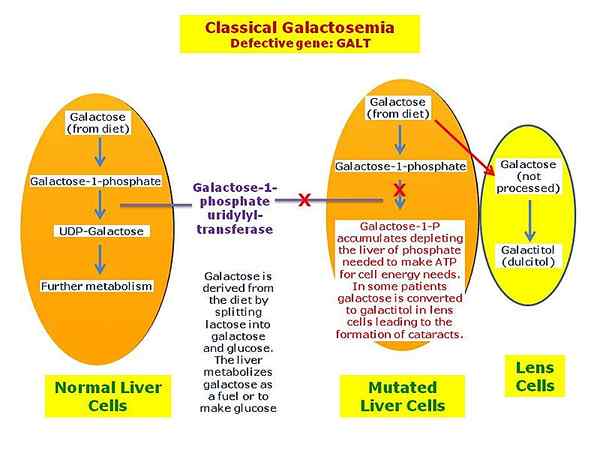
Galactosämie ist eine seltene ererbte Stoffwechselstörung, die aufgrund der Akkumulation von Galaktose-verwandten Chemikalien in verschiedenen Organen des Körpers auftritt. Es ist ein seltener angeborener Stoffwechselfehler, der die Fähigkeit eines Individuums beeinflusst, Galactose richtig zu metabolisieren. Galactosämie folgt dem autosomal rezessiven Vererbungsmodus und ist auf ein Enzym zurückzuführen, das für einen angemessenen Galaktoseabbau verantwortlich ist. Diese Störung wird durch einen Mangel eines Enzyms namens Galactose 1 Phosphat Uridylyltransferase (GALT) verursacht und ist auf Mutationen in Genen zurückzuführen Galt, Galk1 Und STURM.
Abbildung 02: Galactosämie
Die Symptome von Galactosämie können Krämpfe, Reizbarkeit, Lethargie, schlechte Fütterung, schlechte Gewichtszunahme, gelbe Haut, weiße Augen (Gelbsucht) und Erbrechen, Durchfall, Katarakte, Leberschäden, Nierenprobleme, Entwicklungsstörungen und Eierstöcke bei Mädchen umfassen. Darüber hinaus kann die Diagnose dieser Erkrankung durch Familienanamnese, Blutuntersuchungen, Urintests, DNA -Analyse und Enzymanalyse durchgeführt werden. Darüber hinaus umfasst die Behandlungsoption für Galactosämie eine niedrige Galactose -Diät. Dies bedeutet, dass Milch und andere Lebensmittel, die Lactose oder Galactose enthalten. Darüber hinaus kann die Sprachtherapie, individuelle Bildungsplanung und Intervention sowie die Hormonersatztherapie nützlich sein, um andere Symptome zu behandeln.
Was sind die Ähnlichkeiten zwischen Phenylketonurie und Galaktosämie?
- Phenylketonurie und Galaktosämie sind zwei ererbte Stoffwechselstörungen oder angeborene Stoffwechselfehler.
- Beide sind auf ererbte genetische Mutationen zurückzuführen, die Enzyme verursachen.
- Beide folgen dem autosomal rezessiven Vererbungsmodus.
- Sie können durch Einschränkung von Diäten behandelt werden.
Was ist der Unterschied zwischen Phenylketonurie und Galactosämie?
Phenylketonurie ist eine seltene ererbte Stoffwechselstörung, die aufgrund der Ansammlung einer Aminosäure verursacht wird, die Phenylalanin in verschiedenen Körperorganen namens. Dies ist daher der Hauptunterschied zwischen Phenylketonurie und Galactosämie. Darüber hinaus ist Phenylketonurie auf die Mutation des PAH -Gens zurückzuführen, während Galactosämie auf die Mutationen von Genen wie zurückzuführen ist Galt, Galk1, Und STURM.
Die folgende Infografik zeigt die Unterschiede zwischen Phenylketonurie und Galactosämie in tabellarischer Form für Seite für Seitenvergleich.
Zusammenfassung - Phenylketonurie gegen Galactosämie
Phenylketonurie und Galaktosämie sind zwei ererbte Stoffwechselstörungen oder angeborene Stoffwechselfehler. Phenylketonurie tritt aufgrund der Akkumulation einer Aminosäure auf, die Phenylalanin in verschiedenen Körperorganen namens. Dies fasst den Unterschied zwischen Phenylketonurie und Galactosämie zusammen.
Referenz:
1. „Phenylketonurie.Nord (Nationale Organisation für seltene Störungen).
2.„Galactosämie: Symptome, Ursachen, Diagnose, Behandlung.”WebMD.
Bild mit freundlicher Genehmigung:
1. „Phenylalanin-Metabolismus“ von Allen Gathman (CC BY-NC-SA 2.0) über Flickr
2. "Galactosämie" durch Störungen (CC BY-SA 4.0) über Commons Wikimedia