Was ist der Unterschied zwischen Alkaptonurie und Phenylketonurie
Der Schlüsselunterschied zwischen Alkaptonurie und Phenylketonurie ist, dass Alkaptonurie eine ererbte genetische Störung ist, die sich aus der Unfähigkeit ergibt.
Inbornfehler des Stoffwechsels sind seltene ererbte genetische Erkrankungen. Unter diesen Bedingungen kann der Körper Nahrung nicht richtig in Energie verwandeln. Die Erkrankungen der angeborenen Stoffwechselstörungen werden normalerweise aufgrund von Defekten in bestimmten Enzymen verursacht, die dazu beitragen, Teile der Nahrung abzubauen. Es gibt viele verschiedene Arten von angeborenen Stoffwechselfehlern. Einige von ihnen sind Fructose -Intoleranz, Galaktosämie, Urinkrankheit von Ahornzucker, Alkaptonurie und Phenylketonurie.
INHALT
1. Überblick und wichtiger Unterschied
2. Was ist Alkaptonurie
3. Was ist Phenylketonurie
4. Ähnlichkeiten -Alkaptonurie und Phenylketonurie
5. Alkaptonurie gegen Phenylketonurie in tabellarischer Form
6. Zusammenfassung -Alkaptonurie gegen Phenylketonurie
Was ist Alkaptonurie?
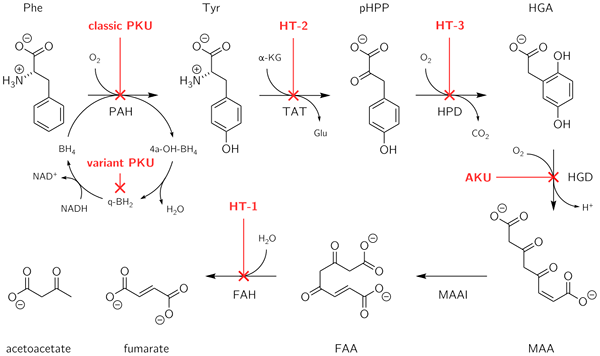
Alkaptonurie ist eine ererbte genetische Störung, die sich aus der Unfähigkeit ergibt, zwei Aminosäuren zu metabolisieren: Tyrosin und Phenylalanin. Es ist eine Art angeborener Stoffwechselfehler. Diese Störung ist auf eine Mutation in einem Gen genannt HGD, Das ist verantwortlich für die Herstellung eines als HGD namens HGD (Homogentisate 1, 2-Dioxygenase). Das HGD -Enzym ist am Metabolismus des aromatischen Aminosäuren Tyrosin und Phenylalanin beteiligt. Menschen mit HGD-Mutation können Homogentisäure nicht metabolisieren, die aus Tyrosin in 4-Maleylacetoacetat erzeugt werden. Dieser Defekt führt zur Akkumulation von Homogentisinsäure in Blut und Geweben. Darüber hinaus werden in diesem Zustand Homogentisinsäure und ihre oxidierte Form (Alkapton) im Urin ausgeschieden, was dem Urin eine ungewöhnlich dunkle Farbe verleiht.
Abbildung 01: Alkaptonurie
Das Zeichen und die Symptome umfassen dunkel gefärbte Gewebe im Körper, Gelenk- und Knochenprobleme (Arthrose), Verdickung und blauschwarze Verfärbung des Ohrknorpels (Ochronose), schwarz oder rötlich-braun Augen, verfärbter Schweiß, blau oder schwarz gesprenkelte Bereiche mit Haut, bläuliche Farbnägel, Atemschwierigkeiten, Herzprobleme (Hardenklappen und steife Blutgefäße), Nierensteine, Blasensteine und Prostata -Steine. Die Diagnose dieser Erkrankung kann durch körperliche Untersuchung, detaillierte Patientenanamnese, Urintest, Gaschromatographie zum Testen von Homogentisäure und DNA -Test zur Überprüfung der Mutation des HGD -Gens gestellt werden. Zu den Behandlungsoptionen gehört auch die Angabe von Nisiton zur Reduzierung der Homogentisinsäure im Körper, die Einschränkung der Proteine in der Ernährung, das Training für Schmerzen und Steifheit und Schmerzlinderung durch Schmerzmittel.
Was ist Phenylketonurie?
Phenylketonurie ist eine ererbte genetische Störung, die sich aus der Unfähigkeit ergibt, nur eine Aminosäure zu metabolisieren: Phenylalanin. Diese Erkrankung ist normalerweise auf eine Mutation in einem Gen zurückzuführen, das als bekannt ist PAH, die für ein Enzym kodiert, das als Phenylalaninhydroxylase bekannt ist. Dieses Enzym ist notwendig, um Aminosäurephenylalanin auf das Aminosäuretyrosin zu metabolisieren. Wenn sich die Phenylalaninhydroxylase -Aktivität durch Mutation verringert, akkuliert sich Phenylalanin und wird in Phenylpyruvat (Phenylketon) umgewandelt, das im Urin nachgewiesen werden kann.
Abbildung 02: Phenylketonurie
Das Zeichen und die Symptome dieser Störung können muffige Geruch in Schweiß, Haut oder Urin, neurologische Probleme, einschließlich Anfälle, helle Haut und blaue Augen, abnormal kleiner Kopf, Hyperaktivität, intellektuelle Behinderung, verzögerte Entwicklung, Verhalten, emotionaler, sozialer Probleme und umfassende Probleme umfassen psychische Störungen. Die Diagnose wird durch Neugeborenen -Blutuntersuchungen, klinische Bewertung und DNA -Tests auf Genmutation durchgeführt. Darüber hinaus erfolgt die Behandlung hauptsächlich durch die Regulierung der Ernährung, die Lebensmittel umfasst, die niedrige Phenylalaninspiegel (Einschränkungsproteine) und eine Sonderformel für Babys mit einer geringen Menge Muttermilch enthalten. Das Medikamenten -Saproperin -Dihydrochlorid kann in einigen Fällen auch nützlich sein. Dieses Medikament ist ein Cofaktor für das Enzym Phenylalaninhydroxylase, das seine Aktivität verstärkt.
Was sind die Ähnlichkeiten zwischen Alkaptonurie und Phenylketonurie?
- Alkaptonurie und Phenylketonurie sind zwei angeborene Stoffwechselfehler.
- Beide sind ererbte genetische Störungen.
- Diese Störungen folgen autosomal rezessiven Vererbung.
- Beide Störungen führen zur Akkumulation von Metaboliten in Körpergeweben.
- Sie werden hauptsächlich durch Einschränkung von Diäten auf hoher Proteinbasis behandelt.
Was ist der Unterschied zwischen Alkaptonurie und Phenylketonurie?
Alkaptonurie ist eine ererbte genetische Störung, die sich aus der Unfähigkeit ergibt, die beiden Aminosäuren, Tyrosin und Phenylalanin. Dies ist daher der Hauptunterschied zwischen Alkaptonurie und Phenylketonurie. Darüber hinaus beträgt die globale Prävalenz von Alkaptonuria 1 in 250000 bis 1000000 Lebendgeburten, während die globale Phenylketonurie -Prävalenz 1 in 23930 Lebendgeburten beträgt.
Die folgende Infografik stellt die Unterschiede zwischen Alkaptonurie und Phenylketonurie in tabellarischer Form für Seite und Seitenvergleich dar.
Zusammenfassung -Alkaptonurie gegen Phenylketonurie
Inbornfehler des Stoffwechsels sind seltene ererbte genetische Erkrankungen. Alkaptonurie und Phenylketonurie sind zwei angeborene Stoffwechselfehler. Alkaptonurie resultiert aus der Unfähigkeit, zwei Aminosäuren Tyrosin und Phenylalanin zu metabolisieren, während Phenylketonurie die Unfähigkeit resultiert, das Aminosäure -Phenylalanin zu metabolisieren. Dies fasst den Unterschied zwischen Alkaptonurie und Phenylketonurie zusammen.
Referenz:
1. „Alkaptonurie.NHS -Auswahl, NHS.
2. „Phenylketonurie (PKU).Mayo Clinic, Mayo Foundation für medizinische Ausbildung und Forschung.
Bild mit freundlicher Genehmigung:
1. "Ochronose" von Universidad CES - (CC von 3.0) über Commons Wikimedia
2. "Angeborene Fehler des Stoffwechsels von Phenylalanin und Tyrosin" von Bradford Morris - eigene Arbeit (CC BY -SA 4).0) über Commons Wikimedia